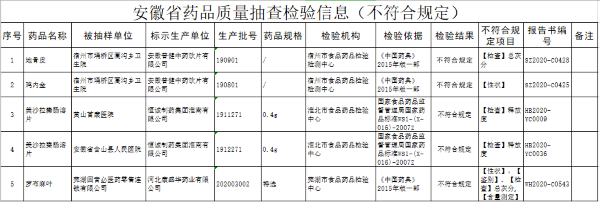
医疗AI企业闯关三类证 尚未有1家完成临床试验
[2018年8月1日起正式施行的《医疗器械分类目录》(下称《目录》)规定,医疗人工智能产品要比照《目录》中的“医用软件”子类,申办医疗器械许可证。对于医疗AI企业来说,产品要想顺利实现商业化,必须“持证上岗”。然而,至今尚未有任何一家医疗AI企业完成临床试验。]
[按照医疗AI产品一般性规定的病例数,如果全部做前瞻性研究,仅临床试验的费用大约在1000万。如果像数坤科技做混合型,全部的试验费用也接近千万。]
能不能拿到一张医疗器械许可证,是所有医疗AI企业能否进入下一赛道的衡量标准。
2018年8月1日起正式施行的《医疗器械分类目录》(下称《目录》)规定,医疗人工智能产品要比照《目录》中的“医用软件”子类,申办医疗器械许可证。对于医疗AI企业来说,产品要想顺利实现商业化,必须“持证上岗”。
然而,至今尚未有任何一家医疗AI企业完成临床试验。
闯关三类证
《目录》指出,医用软件按照预期用途分为辅助诊断类和治疗类。诊断功能软件风险程度不仅依据处理对象(如癌症、恶性肿瘤等疾病的影像)为判定依据,还按照其采用算法的风险程度、成熟程度、公开程度等为判定依据。
根据《目录》,如果医疗AI产品通过算法提供诊断建议,仅仅辅助诊断而不直接给出诊断结论,按照第二类医疗器械管理。如果医疗AI产品通过其算法对病变部位进行自动识别,并提供明确的诊断提示,相比较而言风险级别较高,则按照第三类医疗器械管理。业界将相应类别的医疗AI产品分别称为“二类AI”和“三类AI”。
第一财经记者注意到,“二类AI”可直接在省级药监局申请,“三类AI”则必须经国家药监局审批,并且必须做临床试验。
上海医疗器械行业协会法律咨询顾问岳伟对第一财经记者分析,《目录》中没有“智能医疗器械”的分类,强调的是“独立软件医疗产品”。医疗器械的分类根据风险程度、对人体的作用部位和侵入程度来决定,“智能化”产品的智能程度越高,可能产生的风险越大,分类的等级就高。如果用软件进行“测量、计算、提示”,最后由医生进行处理,风险就低,可能是二类产品;如果用软件进行“诊断、治疗、给药”,直接替代医生的部分工作,风险就高,而现代AI技术一般不会再做低等简单的工作,所以真正称得上AI的产品,大部分是三类产品。
“现代医疗产品都用计算机系统、都会附带不同的软件,以前有些智能化产品只是表面附有一定软件处理功能,而现在讨论的是具有人工智能处理临床预期用途的独立软件产品,目前《目录》中并没有给出AI医疗产品的定义,所以这里的结果、变化、空间非常大。”岳伟说。
从《目录》的分类来看,市场上大部分医疗AI产品应属于三类医疗器械。根据蛋壳研究院发布的《2018医疗人工智能报告》(下称《报告》),为应对这一政策,大部分企业采取增删诊断功能的办法,将产品同时申报二、三类器械。目前,包括推想、深睿、Airdoc等在内的多家企业已经率先获得二类证书,汇医慧影、图玛深维、推想、深睿、Airdoc、依图医疗、神州德信等AI企业都在着手进行三类医疗器械的申报。
临床试验成本高昂
《报告》显示,按照医疗器械注册流程,产品从申报到最终过审要经过产品定型、检测、临床试验、注册申报、技术审评、行政审批六步,必须完成前一步骤才可进行下一步。
数坤科技创始人及CEO马春娥告诉第一财经记者,包括数坤科技在内、目前申报三类器械许可证的产品全都停留在临床试验阶段,还未有任何一款产品进行到注册申报阶段。检测报告和临床试验报告是注册申报的前提条件,通过之后国家药监局才会正式受理。
2018年9月,数坤科技的冠心病AI产品已经完成多中心验证(多位研究者按同一试验方案在不同地点和单位同时进行的临床试验,各中心同期开始与结束试验,由一位主要研究者总负责)。多中心验证相当于前期研究,目的在于验证产品的有效性和安全性,通过后才能进行国家药监局要求做的临床试验。
医学上,临床试验分为前瞻性临床试验(根据方案招募受试者,按方案治疗或观察病例)和回顾性临床试验(收集已经发生的符合方案条件的病例信息),马春娥表示,前者大约需要1年半到2年的时间,后者1年左右能完成,数坤科技选择的是前瞻性和回顾性混合的方案。“因为AI产品验证要求的病例量特别大,如果全都做前瞻性,时间太长。在实际操作层面上,药监局那边也同意以回顾性病人为主,补充一些前瞻性病人的混合方案。”
前期验证和临床试验的费用各在300万~500万,而临床试验的前瞻性病人越多,费用越高。按照医疗AI产品一般性规定的病例数,如果全部做前瞻性研究,仅临床试验的费用大约在1000万。如果像数坤科技做混合型,全部的试验费用也接近千万。
Airdoc是国内第一家申报三类证书的医疗AI企业,送检了中国第一台装载待检人工智能AI软件的服务器。在2018年11月拿到二类证后,时任Airdoc副总裁的张京雷曾向第一财经记者表示,三类医疗器械要做临床试验,大概需要2~3年时间。
审批流程和要点尚未出台
2018年12月,国家药监局在北京举办了一场专项公益培训会,介绍了三类医疗AI的审批流程和审批要点,以及临床试验的要求建议等。岳伟表示,这些审批流程和要点不是法律法规,是技术文件,“技术文件可以根据技术的发展和产品的特点变化而改变”。
第一财经记者在采访中了解到,目前已经有90多家企业跟国药监就三类证的申请进行沟通,具体包括数据集的整理、敏感性特异性指标的评估、安全性有效性的评估等方面。
马春娥告诉记者,国药监跟美国FDA(FoodandDrugAdministration,食品药品管理局)沟通紧密,亦参考了美国的做法,目前已经初步形成统一标准,前述公益培训会的召开是给所有企业一个导引。
公开资料显示,FDA根据设备的可能使用及其给病人带来的风险分三类监管医疗设备:第一类是低风险设备,如医用手套,第二类是中等风险设备,如CT机,第三类是最高风险设备,如支架。FDA的AI影像系统分为两种:Computer-AidedDetection(简称CADe)和Computer-AssistedDiagnostic(简称CADx),前者用于检测身体异常状况,后者评估疾病的存在与否,比如严重性、疾病分类或预测。
为了加快医疗AI审评审批进程,2017年7月,FDA发布数字健康创新行动计划(DigitalHealthInnovationActionPlan,简称DHIAP),考虑到产品的特征、临床前景、独特的用户界面以及行业内引进此类产品的商业周期等,专门针对电子健康产品建立了新的、实效性强的监管方法。
美国的医疗AI审批快于中国:2018年2月,FDA审批了第一个针对中风的AI诊断决策支持产品——Viz.AI的ContaCT;同月审批了第一个针对儿童自闭症的AI诊断决策支持系统——Cognoa公司的一款深度学习应用;2018年4月,批准通过首个应用于一线医疗的自主式人工智能诊断设备——IDx-DR的软件程序;2018年5月,批准了一款名为OsteoDetect的新型人工智能工具,该工具是一种计算机辅助检测和诊断软件应用程序,使用AI算法来帮助医生以更快的速度诊断腕骨骨折。
马春娥表示,相比国内绝大部分医疗AI产品被定为三类,大部分AI在美国都被定为二类,国内对待医疗AI产品的审批态度更慎重。
岳伟认为,国内AI医疗产品获得注册证的困难之处在于:对产品的认识速度跟不上审评的认识程度,对高风险产品的临床验证满足不了临床的实际需求。而这些问题的解决还有赖技术研究的发展和临床验证的积累,只是在激烈的竞争中,企业的时间和费用成本都很昂贵。